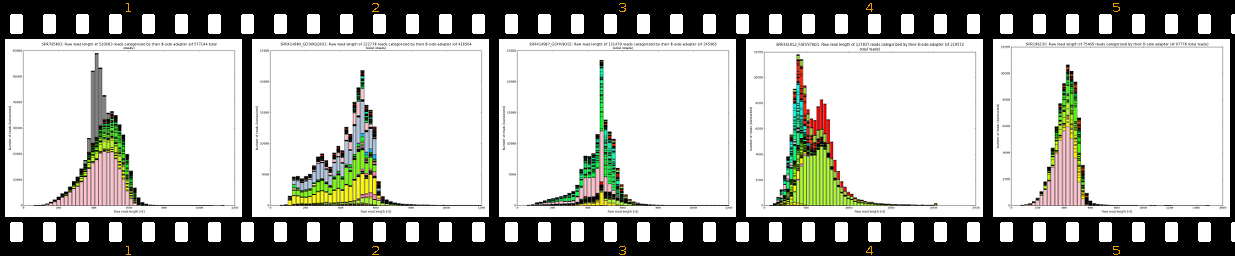
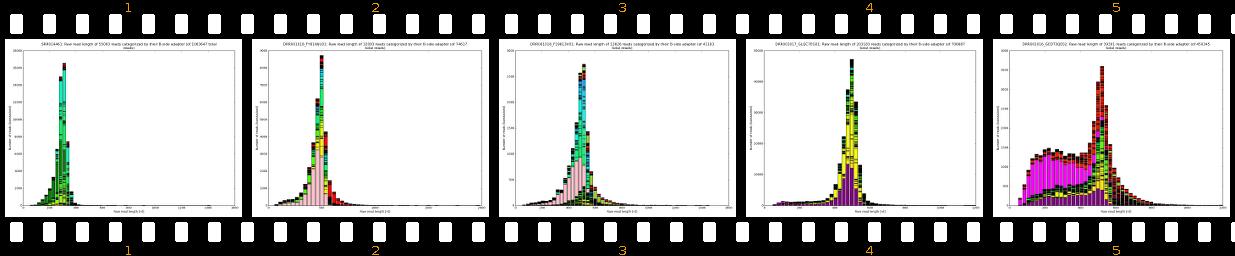
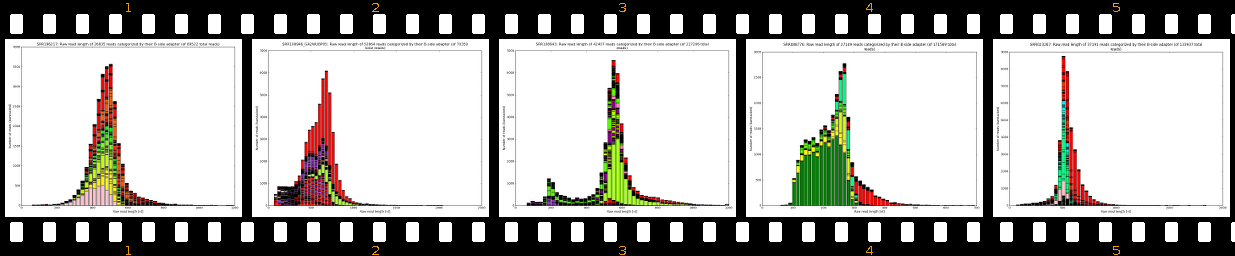
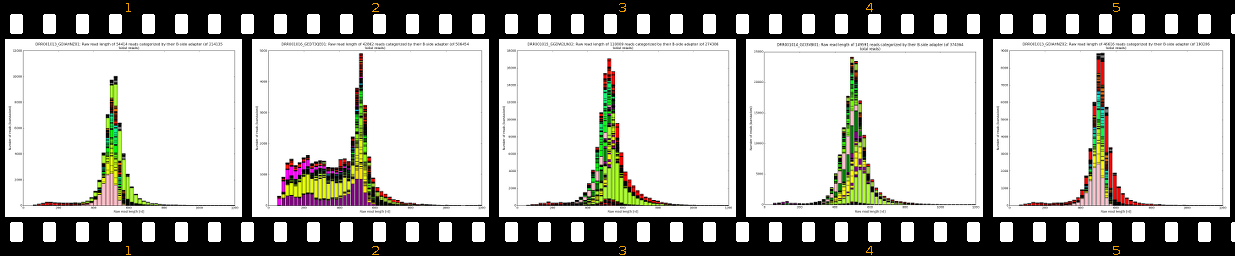
Our software pipeline is able to correct data from any Roche 454TM Life SciencesTM sequencing machine (GS20TM, GS FLXTM, GS FLX+TM, GS JuniorTM). Also some datasets from other sequencing technologies can be processed and fixed (Illumina, IonTorrent). Please see Supported protocols for details. The core analysis package includes inspection of raw reads, evaluation of adapter localization and orientation followed by their precise removal along with MIDs, chimeras, software/virtual junk sequences and physical artifacts. The cleanup results in improved assemblies (less chimeras, less but larger both contigs and scaffolds) and also in some cases in much faster assembly times. Extra-paid result files allow scientists to evaluate sample preparation quality, sequencing quality and importantly, provide basis for successful troubleshooting.




















The key features of the software solution are:
- supports probably all ever reported experimental setups (amplicon, shotgun, transcriptome, all types of MIDs from Roche, even mixes of samples prepared via different protocol sequenced within physically same sequencing region, some funny, likely mis-caried experimental designs)
- detection and removal of MID (Multiplex IDentifier) tags from both ends (Roche software still cannot do that)
- detection and removal of thousands of Roche and 3rd-party adapters (dynamically generated queries specific for each individual dataset)
- detection and removal of thousands of known chimeras and other artifacts isolated from over 2220 of publicly available datasets
- matched regions are aligned to flows (the only way to achieve exact trimming)
- outputs corrected data in SFF / FASTA+QUAL / FASTQ file formats
- adapters/MIDs are annotated in the SFF / CSV files and prevented to interfere with downstream processing
- authentic polyA-tails of transcripts are corrected in FASTQ / FASTA+QUAL files and retained to ensure the best assembly results (reconstruct full-length mRNA sequences from pyrosequencing reads)
- junk sequences are unleashed in raw data, annotated in output SFF file and omitted from cleaned data files (faster and better assemblies)
- get longer reads by rescuing carefully selected portions of the sequence which Roche sacrificed in the name of “low-quality” for no good reason (get better assemblies with even less scaffolds, contigs, gaps with increased scaffold/contig lengths and coverage)
- rescue the second mate to yield even doubled number of paired-end reads compared to original
- reduce number of scaffold contigs by half to 1/4
- increase scaffold length by 30%
- transform e.g. 3 scaffolds into just one
- reports overall sequencing performance in CSV file format for your own Excel spreadsheet-based processing
- determine your sequencing overhead
- learn how many nucleotides were wasted in sequencing adapters/artifacts introduced by certain molecular-biology protocols
- learn how much sequence is sacrificed in unused portions of your reads (your room to improve)
- learn your effective sample insert size distribution and compare it to e.g. Agilent Bioanalyzer traces
- outputs beautiful figures in PNG format for advanced experimental interpretation and troubleshooting